CCSS - Forschungsprojekte der Theoretischen Chemie Arbeitsgruppe Computersimulation
ab initio MD Simulation of Proton Transport in Polymer Electrolytes
Polymer electrolyte membranes provide the environment for proton transport in low temperature fuel cells. Under certain operational conditions, such membranes will develop regions of low water content which are experimentally found to be less conductive than water-rich regions. The motion of protons through narrow necks and pores becomes rate-determining under these conditions. We use the Car-Parrinello ab initio MD simulation method based on quantum-mechanical density functional theory to study proton transport in narrow model pores of a polymer electrolyte such as Nafion in order to understand the mechanism of this transport process such as the amount of water in such pores necessary for efficient proton transfer.
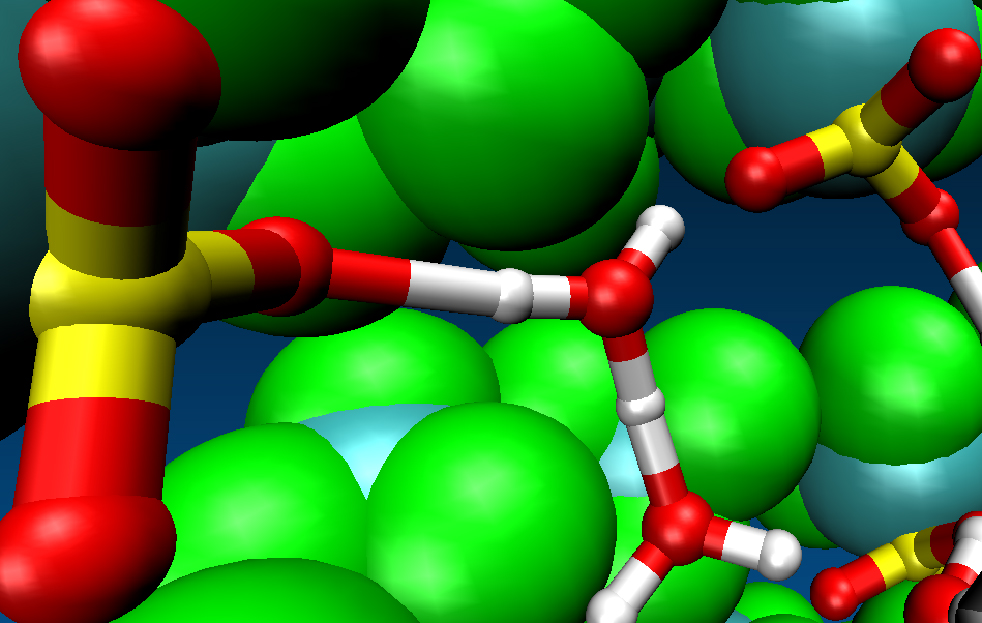
Hydrogen bonded proton cluster in a narrow model pore of a sulfonated perfluoro-polymer electrolyte
Molecular Dynamics of Proton Discharge on Metal Electrodes
Modern particle based computer simulation techniques provide insight into the environment of chemical reactions on the atomic level. Proton transfer in aqueous solutions and proton discharge reactions at metal surfaces belong to a class of reactions where the environment (usually water) is not only a spectator of the reaction but actively participates in it. Reactive trajectory molecular dynamics simulations using reactive force fields are employed to calculate reaction rates for proton discharge as a function of the nature of the metal, electrode potential or surface charge, and the nature of the electrolyte. Thus, atomistic insight into mechanism and driving forces of one of the most basic electrochemical reactions can be gained.
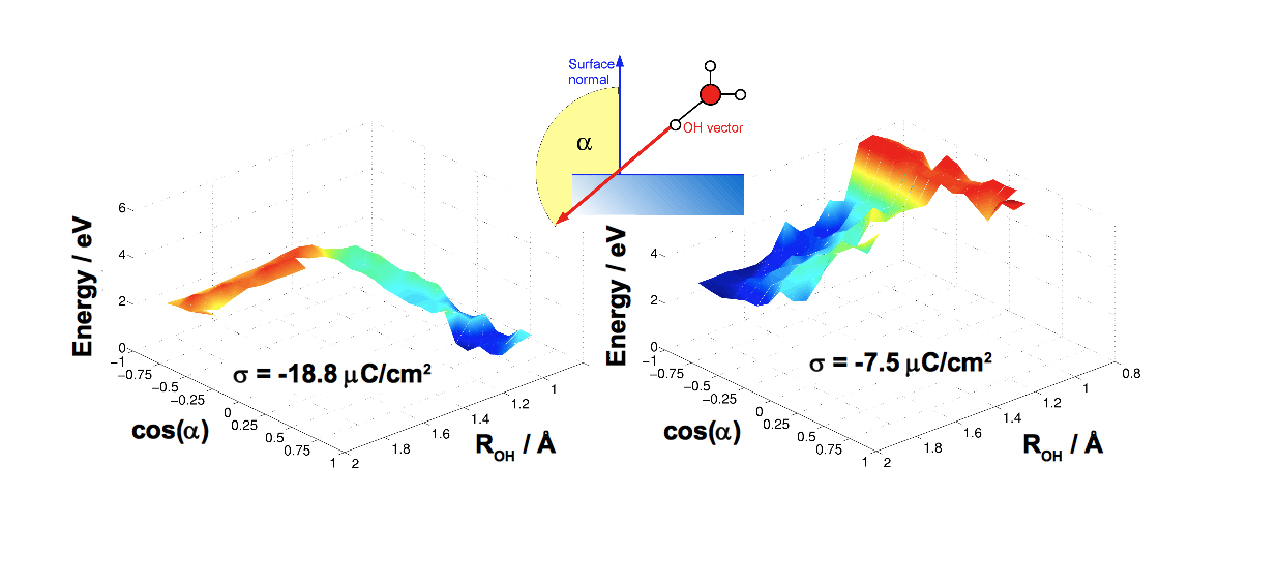
Simulation averaged energy landscape on Pt(111) for low negative (left) and strongly negative (right) electrode charge
Phase Behavior of Thermoresponsive Polymers
PNIPAM (poly-N-isopropylacrylamide) is a thermoresponsive polymer. Above 33° C it undergoes a reversible phase transition from a hydrated swollen state to a dehydrated shrunken state. Individual chains undergo a change from an extended coil to a collapsed polymer. We investigate the phase behavior of this system by molecular dynamics (MD) simulations in the bulk state and on surfaces.
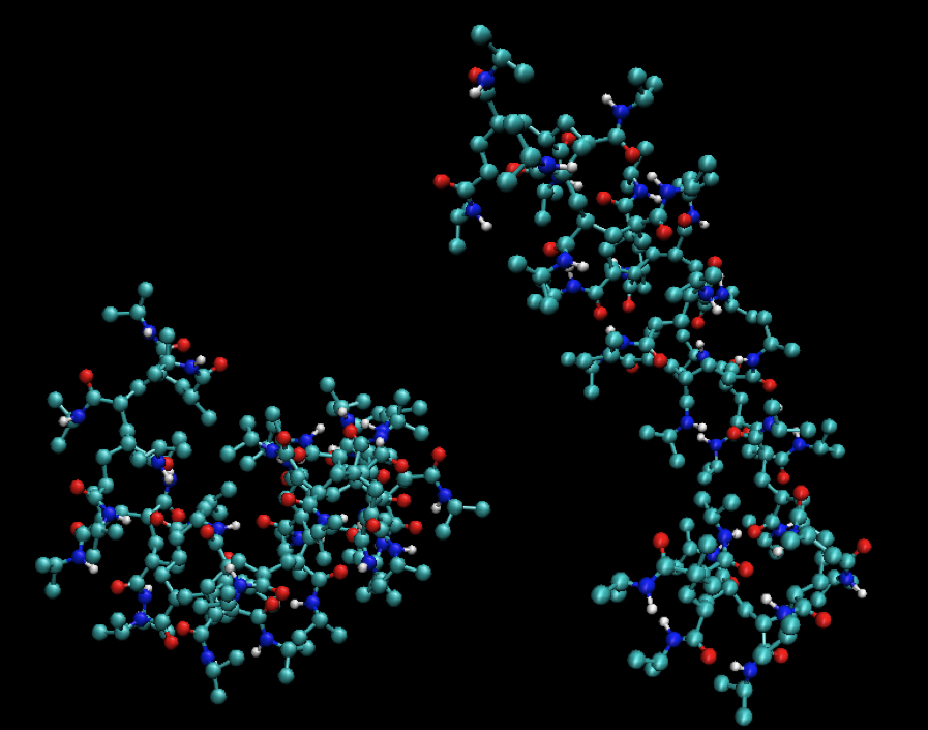
Collapsed and extended conformations of a PNIPAM polymer with 50 repeat units