Research Highlights
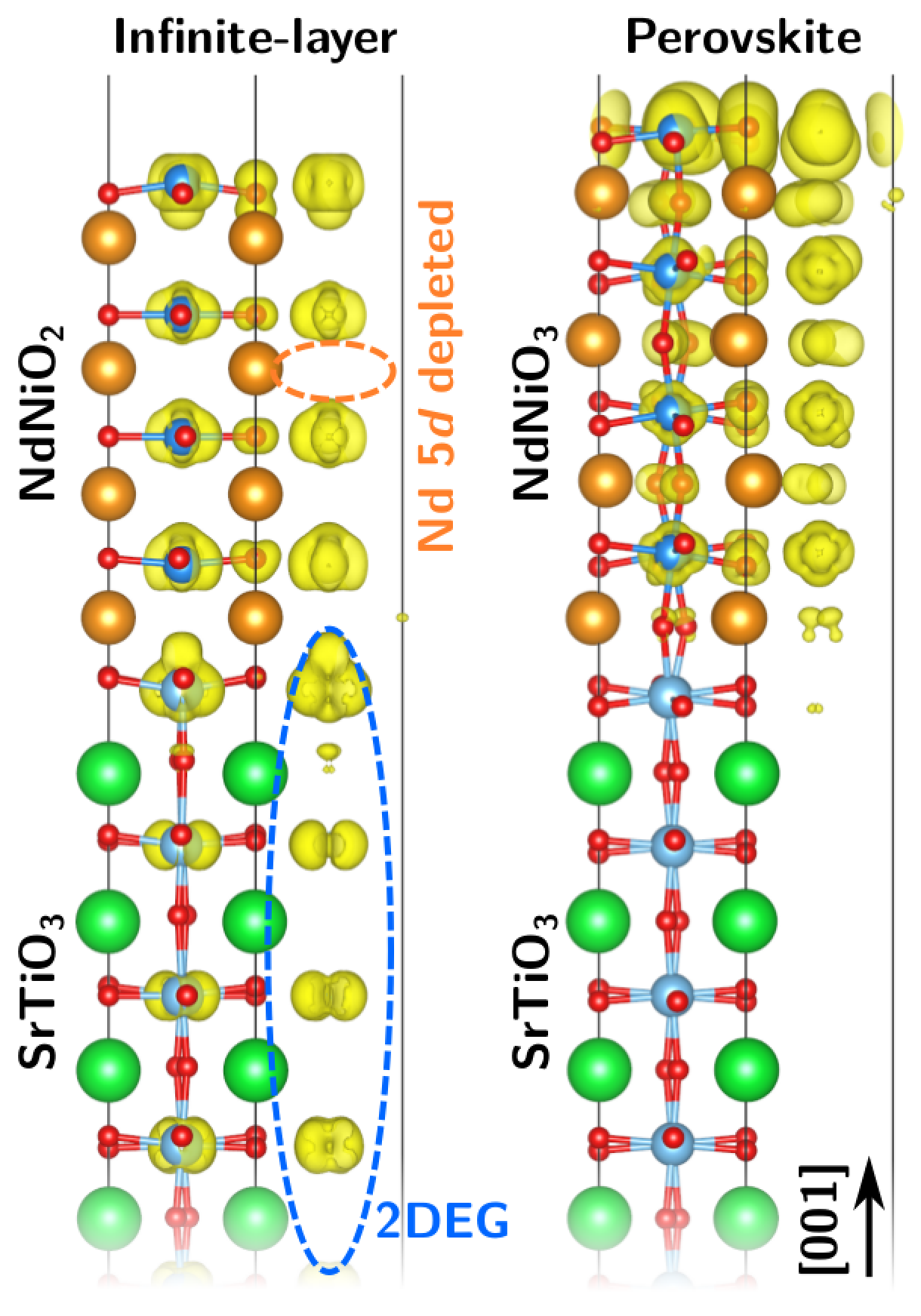
Superconducting nickelatesCuprate-like behavior in a nickel-oxide film
B. Geisler and R. Pentcheva
Phys. Rev. B 102, 020502(R) (2020), Rapid Communication, Editors’ Suggestion
Superconductors transmit electric current without loss at any distance and play an important role in quantum computers and medical imaging. Unfortunately, the stars among the electrical conductors work exclusively at extremely low temperatures. Since the discovery of high-temperature superconducting cuprates with their characteristic copper-oxygen plaquettes in 1986, scientists have been searching for similar behavior in other materials classes. It was not until 2019 that superconductivity was reported in a nickel oxide, but the underlying mechanism is still unclear. Theoretical physicists from the Center for Nanointegration (CENIDE) at the University of Duisburg-Essen (UDE) have therefore studied the electronic properties of the material and found a possible explanation.
Since bulk neodymium nickel oxide (NdNiO2), which exhibits an analogous crystal structure and valence electron number as the cuprates, does not show superconductivity, Prof. Rossitza Pentcheva and Dr. Benjamin Geisler focused on the role of the film geometry. They simulated a 1.5 nanometer thin layer of this so-called infinite-layer nickelate on a strontium titanate substrate (SrTiO3) in comparison to a perovskite (NdNiO3) film based on quantum-mechanical simulations at the supercomputer MagnitUDE.
Two-dimensional electron gas discovered
Despite the fact that both systems have a charge mismatch at the interface, a major difference appears in accommodating it: Only in the infinite-layer case does the charge mismatch lead to the formation of a two-dimensional electron gas at the interface. "It is known from other materials combinations that such a two-dimensional electron gas can be superconducting" explains Pentcheva. Moreover, in contrast to the bulk, the infinite-layer film shows a cuprate-like electronic behavior, indicating that the film geometry may play a significant role in the emergence of superconductivity.
The more that is known about the origin of superconductivity, the better the chances are that the sought-after property can be specifically induced in tailor-made material systems, even at room temperature.
Changing Material Properties by Stretching
Seung Sae Hong, Mingqiang Gu, Manish Verma, Varun Harbola, Bai Yang Wang ,Di Lu , Arturas Vailionis, Yasuyuki Hikita, Rossitza Pentcheva, James M. Rondinelli, Harold Y. Hwang
Unlike ductile metals, oxides are ceramics, known to be brittle and unable to sustain large strains. An international team of scientists, including theoretical physicists from the UDE, were successful in achieving extreme strains of up to 8 percent in oxide membranes. When atoms are pulled away from each other, electrons can localize, thereby leading to unexpected behavior such as a metal-to-insulator transition. This breakthrough, published in "Science”, can serve to selectively design novel functionality in materials – for sensors or detectors, for example. In collaboration with colleagues from Northwestern University (US), Manish Verma and Professor Rossitza Pentcheva have explored the origin and underlying mechanisms for the behavior of lanthanum calcium manganese oxide (La1-xCaxMnO3, LCMO) under extreme strains using large scale density functional theory calculations. The physicists were able to show that while LCMO normally is metallic and ferromagnetic, under tensile strain of 5% it switches to an insulator with antiferromagnetic order, characterized by a stripe-type of charge order of Mn3+ and Mn4+ ions. The statistical distribution of the lanthanum and calcium ions required large simulation cells, the calculations were therefore carried out on UDE's high-performance computer "MagnitUDE".
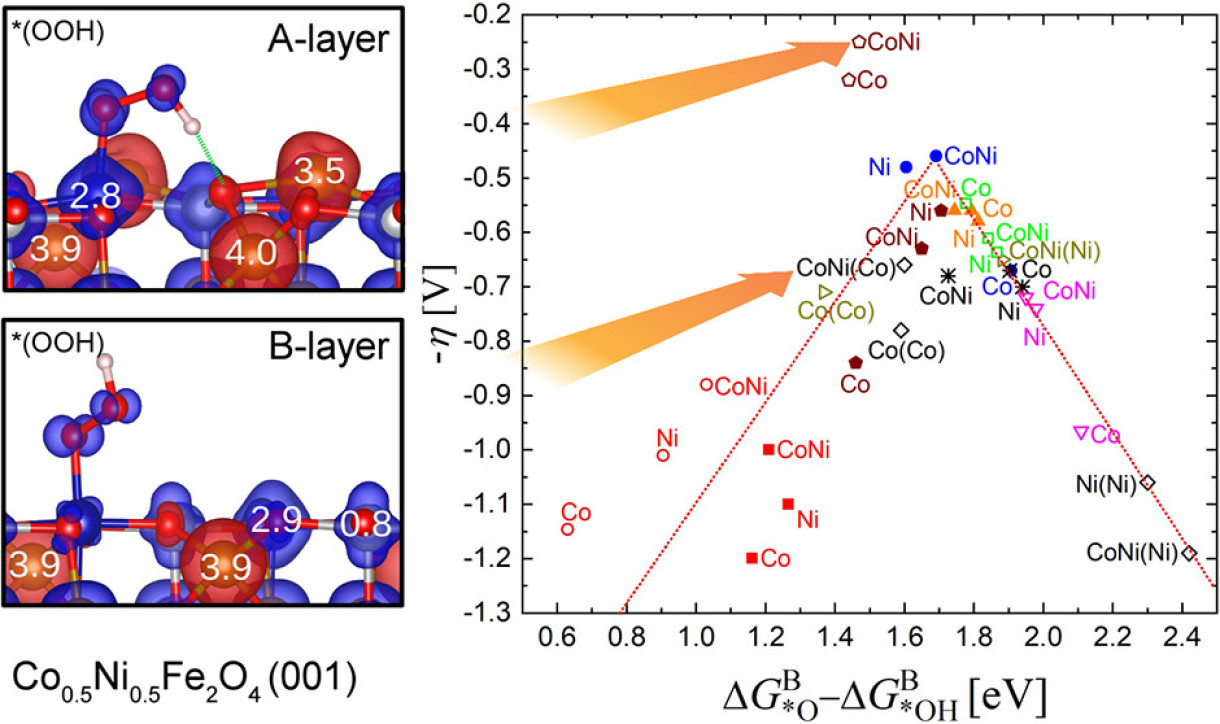
Surface Termination and Composition Control of Activity of the CoxNi1–xFe2O4 (001) Surface for Water Oxidation: Insights from DFT+U Calculations
H. Hajiyani, and R. Pentcheva
ACS Catal. 8, 12,11773-11782 (2018)
Using density functional theory calculations with an on-site Hubbard term (DFT+U), we explore the effect of surface termination and cation substitution on the performance of the CoxNi1–xFe2O4(001) surface (x = 0.0, 0.5, 1.0) as an anode material in the oxygen evolution reaction (OER). Different reaction sites (Fe, Co, Ni, and an oxygen vacancy) were investigated at three terminations: the B-layer with octahedrally coordinated Co/Ni and with an additional half and full monolayer of Fe (0.5A and A-layer, respectively). Ni substitution with an equal concentration of Co and Ni (x = 0.5) reduces the overpotential over the end members for the majority of reaction sites. Surface Co cations are identified as the active sites and the ones at the A-layer termination for x = 0.5 exhibit one of the lowest theoretically reported overpotentials of 0.26 V. The effect of the additional iron layer on the active site modification is 2-fold: analysis of the electronic properties and spin densities indicates that the additional Fe layer stabilizes a bulk-like oxidation state of +2 for Co and Ni at the A-layer termination, whereas at the B-layer termination, they are oxidized to +3. Moreover, the unusual relaxation pattern enables the formation of a hydrogen bond of the OOH intermediate to a neighboring surface oxygen that lowers the reaction free energy of this formerly rate-limiting step, leading to a deviation from the scaling relationship and almost equidistant reaction free-energy steps of intermediates. This renders an example of how a selective surface modification can result in a significant improvement of OER performance.
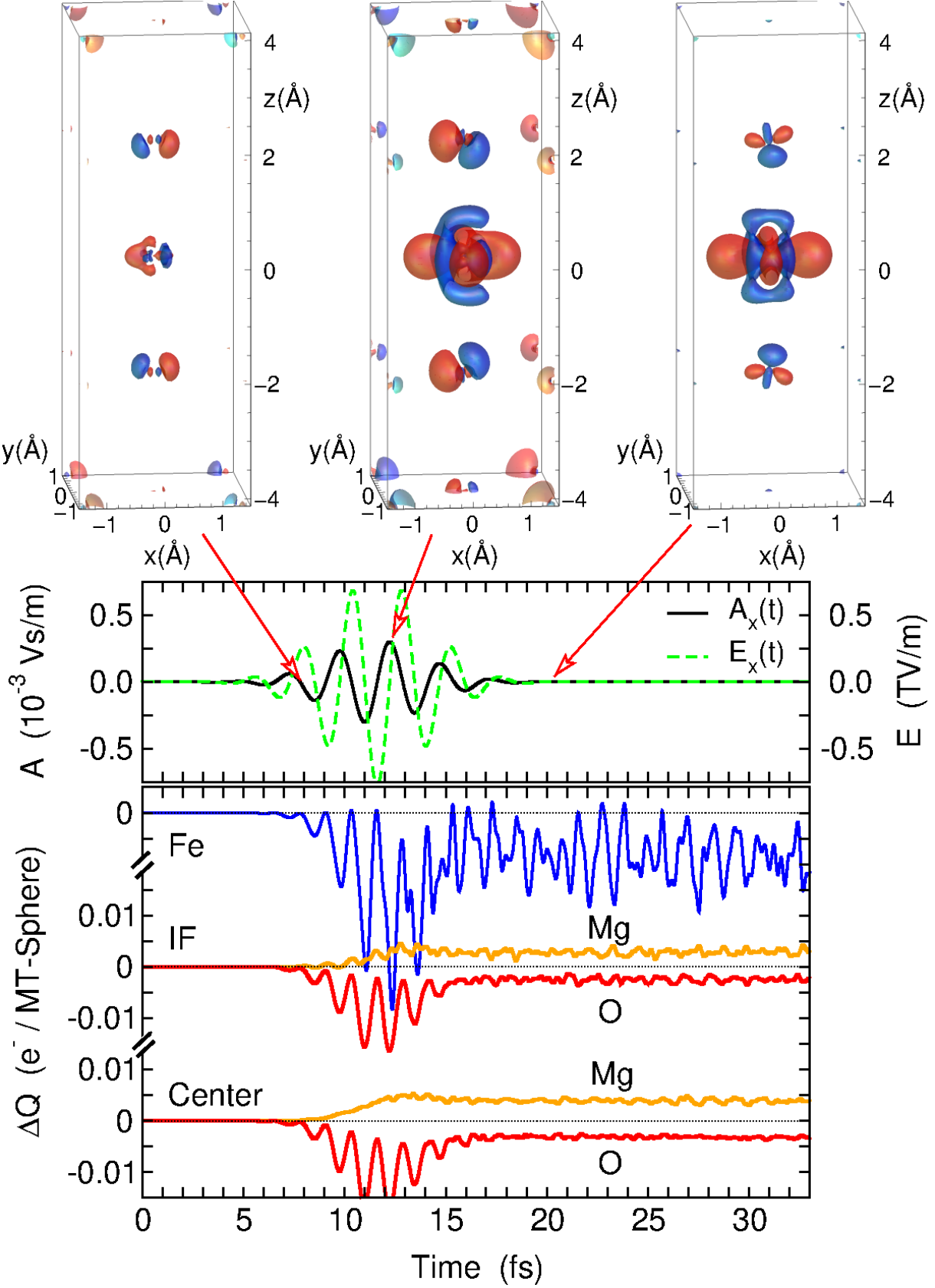
Dynamics of optical excitations in a Fe/MgO(001) heterostructure from time-dependent density functional theory
M. E. Gruner, and R. Pentcheva
Phys. Rev. B 99, 195104 (2019)
In a recent investigation we unraveled the layer-resolved dynamics of excited carriers in a metal/insulator heterostructure with TDDFT in the real-time domain. The figure on the left visualizes the temporal evolution of a Fe1/(MgO)3(001) heterostructure during the excitation with an in-plane polarized laser pulse. The energy of the pulse of 1.63 eV is significantly smaller than the band gap of MgO but reaches the size of the charge transfer gap of the metal/insulator heterostructure. Considerable transient changes occur during the pulse: While the strongest charge redistribution takes place in the Fe layer, a time-dependent change in the occupation is encountered in all layers, mediated by the presence of interface states. The time evolution of the charge density reveals the depletion from in-plane orbitals and accumulation of charge in out-of-plane orbitals. After the pulse, the system reaches a steady state with changes mainly concentrated in the Fe and neighboring MgO layers. Ongoing RT-TDDFT investigations address the evolution of optical excitations in Fe/MgO heterostructures with increased layer-thickness and the extension to other systems with optical anisotropy.
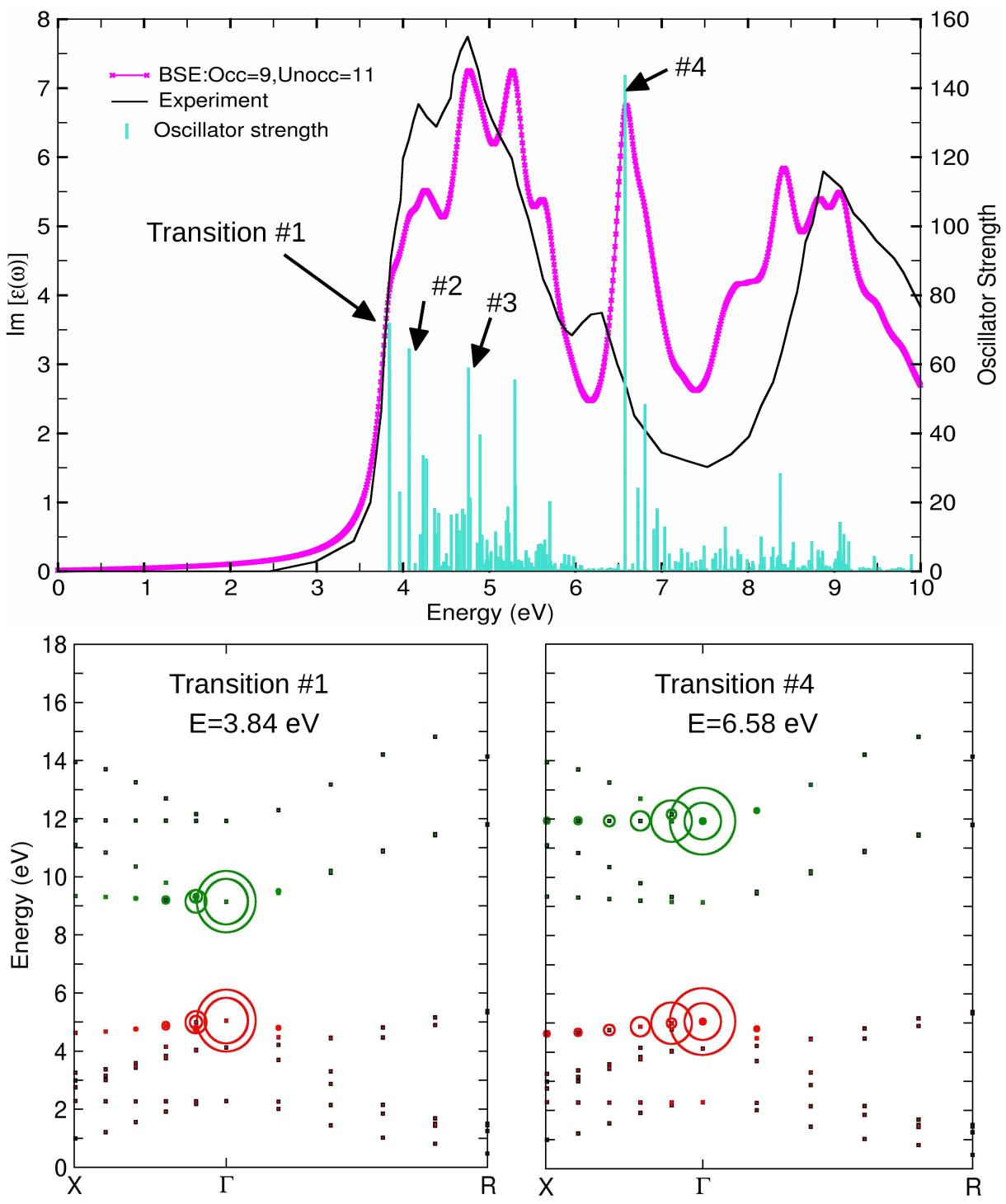
Role of the exchange-correlation functional on the structural, electronic, and optical properties of cubic and tetragonal SrTiO3 including many-body effects
V. Begum, M. E. Gruner, and R. Pentcheva
Phys. Rev. Materials 3, 065004 (2019)
As an important prerequisite for our investigations of transient properties in complex oxide systems, we have systematically explored the structural, electronic, and optical properties of cubic and tetragonal SrTiO3 including many-body effects. Particular focus was laid on the impact of the exchange-correlation functional, by comparing the generalized gradient approximation (PBE96 and PBEsol) and the hybrid functional (HSE06) with the recently introduced strongly constrained and appropriately normed (SCAN) meta-GGA functional, which promises an accuracy comparable to HSE06 at a computational cost comparable to PBE. The figure shows the optical spectrum of cubic SrTiO3 obtained with the SCAN functional , including many body effects in the framework of the GW approximation and excitonic corrections by solving the Bethe-Salpeter equation (BSE) together with the experimental spectrum measured by van Benthem, Elsässer and French, J. Appl. Phys. 90, 6156 (2001) . Strong excitonic effects are found in agreement with previous results and their origin is analyzed based on the contributing interband transitions. The lower panels show the contribution of the excitons at 3.84 eV and 6.58 eV in reciprocal space. The former is localized at G and comprises transition from the top of the valence band (O 2p) to the bottom of the conduction band (Ti t2g), whereas the latter is more delocalized along G -X and involves the Ti eg states. The low dispersion of the contributing bands leads to a high peak in the optical spectrum, which is noticeably affected by the tetragonal distortion (not shown here).
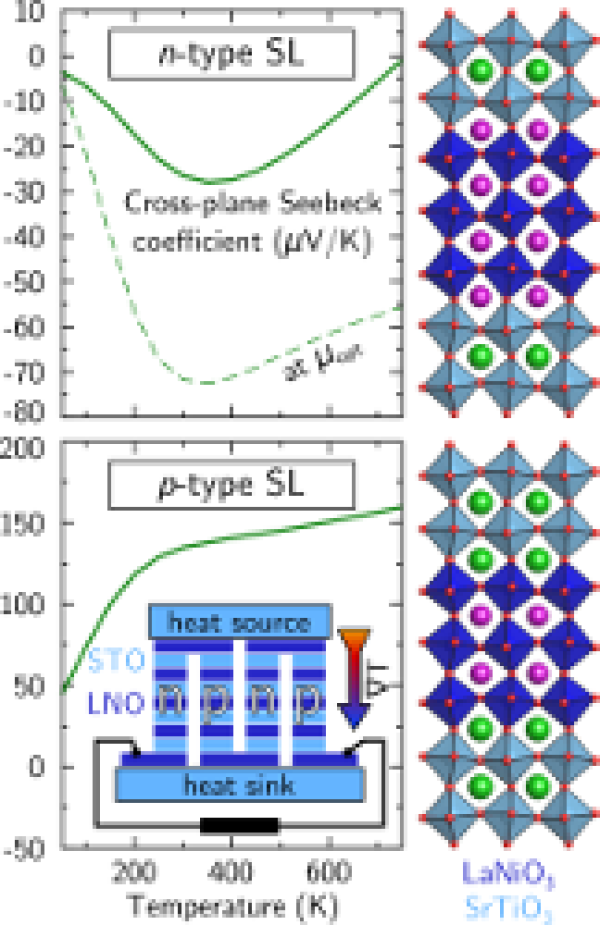
Design of n- and p-type oxide thermoelectrics in LaNiO3/SrTiO3(001) superlattices exploiting interface polarity
B. Geisler, A. Blanca-Romero and R. Pentcheva
Phys. Rev. B 95, 125301 (2017)
We investigate the structural, electronic, transport, and thermoelectric properties of LaNiO3/SrTiO3(001) superlattices containing either exclusively n- or p-type interfaces or coupled interfaces of opposite polarity by using density functional theory calculations with an on-site Coulomb repulsion term. The results show that significant octahedral tilts are induced in the SrTiO3 part of the superlattice. Moreover, the La-Sr distances and Ni-O out-of-plane bond lengths at the interfaces exhibit a distinct variation by about 7% with the sign of the electrostatic doping. In contrast to the much studied LaAlO3/SrTiO3 system, the charge mismatch at the interfaces is exclusively accommodated within the LaNiO3 layers, whereas the interface polarity leads to a band offset and to the formation of an electric field within the coupled superlattice. Features of the electronic structure indicate an orbital-selective quantization of quantum well states. The potential- and confinement-induced multiband splitting results in complex cylindrical Fermi surfaces with a tendency towards nesting that depends on the interface polarity. The analysis of the thermoelectric response reveals a particularly large positive Seebeck coefficient (135 μV/K) and a high figure of merit (0.35) for room-temperature cross-plane transport in the p-type superlattice that is attributed to the participation of the SrTiO3 valence band. Superlattices with either n- or p-type interfaces show cross-plane Seebeck coefficients of opposite sign and thus emerge as a platform to construct an oxide-based thermoelectric generator with structurally and electronically compatible n- and p-type oxide thermoelectrics.
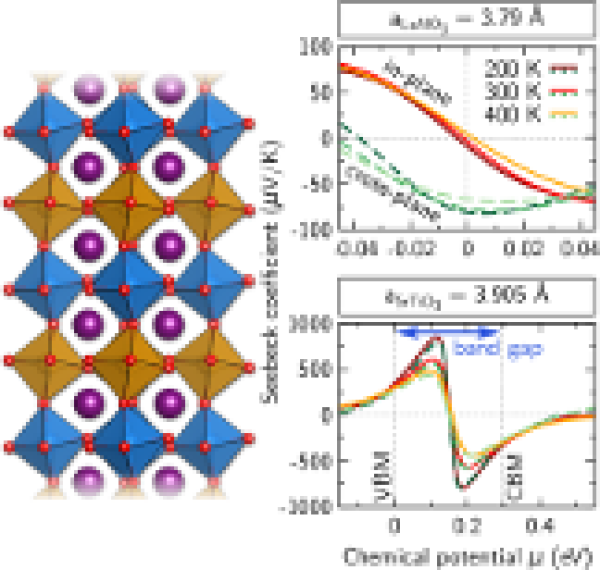
Confinement- and strain-induced enhancement of thermoelectric properties in LaNiO3/LaAlO3(001) superlattices
B. Geisler and R. Pentcheva
Phys. Rev. Materials 2, 055403 (2018),
By combining ab initio simulations including an on-site Coulomb repulsion term and Boltzmann theory, we explore the thermoelectric properties of (LaNiO3)n/(LaAlO3)n(001) superlattices (n=1,3) and identify a strong dependence on confinement, spacer thickness, and epitaxial strain. While the system with n=3 shows modest values of the Seebeck coefficient and power factor, the simultaneous reduction of the LaNiO3 region and the LaAlO3 spacer thickness to single layers results in a strong enhancement, in particular of the in-plane values. This effect can be further tuned by using epitaxial strain as a control parameter: Under tensile strain corresponding to the lateral lattice constant of SrTiO3 we predict in- and cross-plane Seebeck coefficients of ±600 μV/K and an in-plane power factor of 11 μW/K2 cm for an estimated relaxation time of τ=4 fs around room temperature. These values are comparable to some of the best performing oxide systems such as La-doped SrTiO3 or layered cobaltates and are associated with the opening of a small gap (0.29 eV) induced by the concomitant effect of octahedral tilting and Ni-site disproportionation. This establishes oxide superlattices at the verge of a metal-to-insulator transition driven by confinement and strain as promising candidates for thermoelectric materials.
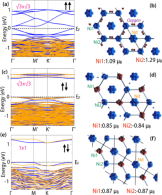
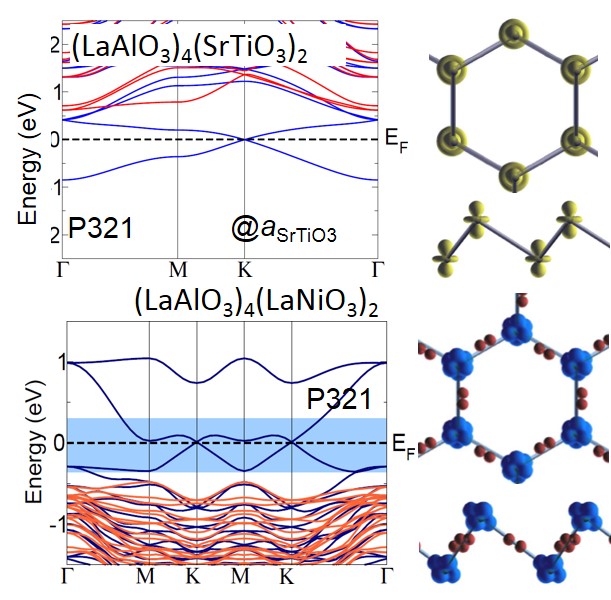
Design of Chern and Mott insulators in buckled 3d oxide honeycomb lattices
D. Doennig, S. Baidya, W. E. Pickett, and R. Pentcheva
Phys. Rev. B 93, 165145 (2016) DOI: 10.1103/PhysRevB.93.165145
Perovskite (LaXO3)2 /(LaAlO3)4 (111) superlattices with X spanning the entire
Mott Electrons in an Artificial Graphenelike Crystal of Rare-Earth Nickelate
S. Middey, D. Meyers, D. Doennig, M. Kareev, X. Liu, Y. Cao, Zhenzhong Yang, Jinan Shi, Lin Gu, P. J. Ryan, R. Pentcheva, J. W. Freeland, and J. Chakhalian
Phys. Rev. Lett. 116, 056801 (2016); DOI: PhysRevLett.116.056801
Deterministic control over the periodic geometrical arrangement of the constituent atoms is the backbone of the material properties, which, along with the interactions, define the electronic and magnetic ground state. Following this notion, a bilayer of a prototypical rare-earth nickelate, NdNiO3, combined with a dielectric spacer, LaAlO3, has been layered along the pseudocubic [111] direction. The resulting artificial graphenelike Mott crystal with magnetic 3d electrons has antiferromagnetic correlations. In addition, a combination of resonant X-ray linear dichroism measurements and ab initio calculations reveal the presence of an ordered orbital pattern, which is unattainable in either bulk nickelates or nickelate based heterostructures grown along the [001] direction. These findings highlight another promising venue towards designing new quantum many-body states by virtue of geometrical engineering.
Control of orbital reconstruction in (LaAlO3)M/(SrTiO3)N(001) quantum wells by strain and confinement
D. Doennig, and R. Pentcheva
Scientific Reports 5, 7909 (2015); DOI: 10.1038/srep07909
DFT+U calculations, performed on (001)-oriented (LAO)M/(STO)N and (NGO)M/(STO)N superlattices with n-type interfaces show a rich set of orbital reconstructions depending on the STO quantum well thickness and c/a ratio. A central finding is the pronounced enhancement of octahedral tilts in the STO quantum well that are not present in the bulk and can be unambiguously associated with the electrostatic doping of the polar n-type interface in these systems. Together with the exotic electronic states found in (111)-oriented (LAO)M/(STO)N SLs, the results demonstrate how strain and the thickness of the STO quantum well can be used to engineer the orbital reconstruction and insulator-to-metal transitions in oxide superlattices.
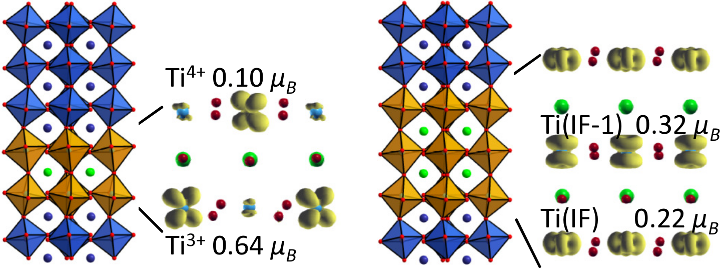
Electrostatic doping as a source for robust ferromagnetism at the interface between antiferromagnetic cobalt oxides
Zi-An Li, N. Fontaíña-Troitiño, A. Kovács, S. Liébana-Viñas, M. Spasova, R. E. Dunin-Borkowski, M. Müller, D. Doennig, R. Pentcheva, M. Farle and V. Salgueiriño
Scientific Reports 5, 7997 (2015); DOI: 10.1038/srep07997
Polar discontinuities at oxide interfaces may be a source of novel functionality which is not available in the bulk constituents. While most of the research so far has focused on heterointerfaces derived from the perovskite structure. H, here we report from high-resolution transmission electron microscopy and quantitative magnetometry a robust – above room temperature (Curie temperature TC>300 K) – stable ferromagnetically coupled interface layer between the antiferromagnetic rocksalt CoO core and a 2–4 nm thick antiferromagnetic spinel Co3O4 surface layer in octahedron-shaped nanocrystals. Density functional theory calculations with an on-site Coulomb repulsion parameter identify the origin of the experimentally observed ferromagnetic phase as a charge transfer process (partial reduction) of Co3+ to Co2+ at the CoO/Co3O4 interface, with Co2+ being in the low spin state, unlike the high spin state of its counterpart in CoO. This finding may serve as a guideline for designing new functional nanomagnets and/or catalysts based on oxidation resistant antiferromagnetic transition metal oxides.
Massive Symmetry Breaking in Quantum Wells: A Three-Orbital Strongly Correlated Generalization of Graphene
D. Doennig, W. E. Pickett, and R. Pentcheva
Physical Review Letters 111, 126804 (2013); DOI: 10.1103/PhysRevLett.111.126804
Physical Review B 89, 121110(R) (2014); DOI: 10.1103/PhysRevB.89.121110
In contrast to the much studied (001)-oriented perovskite superlattices,systems with (111)-orietation promise to host even more exotic electronicstates due to their distinct topology: For example, a bilayer of octahedrally coordianted B-sites of the ABO3 structure forms a buckled honeycomb lattice. Material-specific density functional theory calculations with an on-site Coulomb repulsion term (DFT+U) are used to explore the role of confinement, symmetry breaking, polarity mismatch and strain in the emergence of novel electronic phases. The results illuminate a rich set of competing ground states in polar (LaAlO3)𝑀/(SrTiO3)𝑁(111) and non-polar (LaNiO3)𝑁/ (LaAlO3)𝑀(111) superlattices, ranging from spin-polarized, Dirac-point Fermi surfaces protected by lattice symmetry to charge-ordered Mott or Peierls insulating phases. Analogous to the (001) counterparts, orbital reconstructions and metal-to-insulator transitions depend critically on the thickness of the quantum well 𝑁 and in-plane strain, thus opening avenues for engineering properties at the nanoscale.