Reactivity of distibines and dibismuthines
Reactions with elemental chalcogenes
Distibines and dibismuthines, in which the metal centers adopt the formal oxidation state +II, serves as Lewis bases in reactions with main group metals and transition metals due to the presence of electron lone pairs.[1,2] However, the high s-character of the electron lone pair in dibismuthines render these very weak Lewis bases. In addition, distibines and dibismuthines are promising candidates for redox reactions due to their weak Sb-Sb and Bi-Bi bonds. Insertion reactions with elemental chalcogens yielded R2Sb-E-SbR2 (E = S, Se, Te) and R2Bi-E-BiR2 (E = S, Se, Te).[3,4] Some of these chalcogen-bridged complexes showed thermochromic behavior, pointing to weak intermolecular interactions in the solid state.[5] Unfortunately, their solid state structures were almost unknown. In addition, dichalcogenanes E2R2 (E = S, Se, Te) are known to react with distibines and dibismuthines M2R4 with formation of compounds of the general type R2MER'.[3b,4a,6]]
We recently investigated reactions of distibines Sb2R4 (R = Me, Et) as well as the dibsmuthene Et4Bi2 with elemental chalcogenes (E = S, Se, Te) and reported on the solid state structures of several bis(dialkylstibanyl)- and bis(diethylbismuthanyl)chalcogenanes (R2Sb)2E (R = Me, Et) and (Et2Bi)2E (E = S, Se, Te), which either adopt a syn-syn or syn-anti conformation in the solid state.[7]
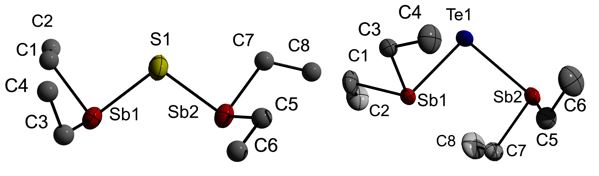
Figure 1: Solid state structures of (Et2Sb)2S and (Et2Sb)2Te.
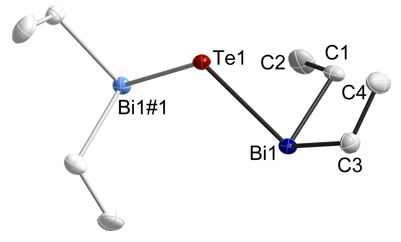
Figure 2: Solid state structures of (Et2Bi)2Te.
The Sb-E-Sb bond and Bi-E-Bi bond angle steadily decreases with increasing atomic number of the chalcogen atom as was expected due to the increasing s-character of the electron lone pairs and the increasing p-character of the E-Bi bonding electron pairs.
The bis(dialkylstibanyl)chalcogenanes show different intermolecular interactions in the solid state. A chain-like structure is observed for the sulfane through the formation of Sb∙∙∙S interactions, in which only one SbEt2 group (Sb1, Sb11) of each independent molecule is involved, while a slightly corrugated ladder-like arrangement wsas observed for the tellurane.
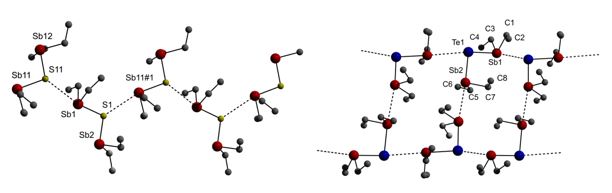
Figure 3: Intermolecular interactions observed for (Et2Sb)2S and (Et2Sb)2Te in the solid state.
The intermolecular interactions can also be discussed in terms of a noncovalent interaction between a covalently-bonded electropositive Sb atom and the lone pair of the Lewis basic chalcogen atom (S, Te). A positive electrostatic potential, referred to as a s-hole, is created on the extension of the covalent bonds at the Sb center, resulting from the anisotropy of the atom’s charge distribution. The s-hole is expected to be rather high at Sb due to its low tendency towards sp-hybridization and its high polarizability.
2. Reactions with dichalcogenes R2E2
Distibines and dibismuthines are also known to react with dichalcogenanes E'2R2 (E' = S, Se, Te).[8,9,10] Compounds of the general type R2SbER' (E = S, Se, Te; R = Me, Et, R' = Me, Ph) were prepared by reaction of distibines Sb2R4 and dichalcogenanes E2R'2 at ambient temperature (Sb2Me4) and -40 °C (Sb2Et4), respectively,[11] by salt elimination reactions[12] and were reported as intermediates in reactions of BiR3 with Te.[13] Some compounds R2SbTeMe (R = Me, Et), which decompose upon heating with formation of the corresponding trialkylstibine SbR3 and RSb(ER')2, were reported to be thermochromic. Me2SbTeMe is a red liquid at room temperature that solidifies at -52 °C with formation of a red solid, which turns yellow-orange at -80 °C. A similar color change was observed when the compound was dissolved in CDCl3. Et2SbTeMe is an orange liquid at room temperature which solidified at -80 °C to give a green-yellow solid. Freezing a solution of this substance in CDCl3 produced a similar change in color.
Even though these types of complexes are well known for years, only limited structural information is available and Ph2BiSePh represents the only structurally characterized compound containing a heavier chalcogen atom.[10e] Surprisingly, no intermolecular contacts were discussed for Ph2BiSePh, even though the closest intermolecular Bi···Se distances (3,897 Å) are below the sum of the van-der-Waals radii (3,97 Å). We recently reported on the single-crystal X-ray structures of Et2SbTeEt and Et2BiTeEt, which were synthesized by reaction of M2Et4 (M = Sb, Bi) and Et2Te2.[14]
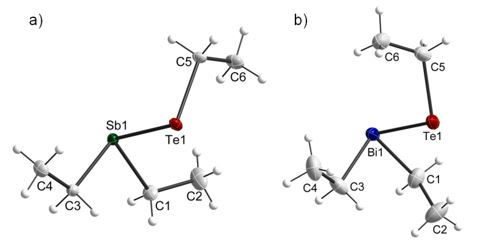
Figure 4: Solid state structure of Et2SbTeEt and Et2BiTeEt, thermal ellipsoids are shown at 50% probability levels.
The Sb-Te bond length (2.7834(4) Å) is shorter than the Bi-Te bond length (2.9116(5) Å) as was expected. The sum of the bond angles at the Sb atom (286.0(3)°) is slightly bigger compared to that at the Bi atom (282.3(8)°), which points to a slightly higher p-character of the bonding electron pairs in Et2BiTeEt.
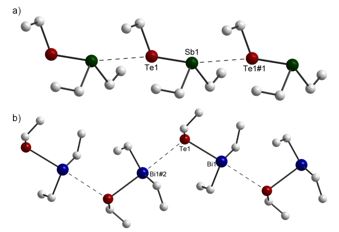
Figure 3: Chains in the packing of Et2SbTeEt (a: formed by translation parallel to a, linear) and Et2BiTeEt (b: formed by 21-axis parallel to b, zig-zag).
Et2SbTeEt and Et2BiTeEt both form chains via intermolecular E···Te interactions (see figure 3). Displaying contact distances well below the sum of the van der Waals radii, they are to be considered attractive from a crystallographic point of view. However, Et2SbTeEt and Et2BiTeEt show different intermolecular interactions, resulting in the formation of almost linear chains (Et2SbTeEt) or zig-zag chains (Et2BiTeEt). According to computational calculations, the small dimeric molecules (Et2SbTeEt)2 and (Et2BiTeEt)2 also prefer linear and zig-zag arrangements in the gas phase.
References
[1] a) Kuczkowski, A.; Schulz, S.; Nieger, M. Angew. Chem. Int. Ed. 2001, 40, 4222; b) Kuczkowski, A.; Schulz, S.; Nieger, M.; Saarenketo, P. Organometallics 2001, 20, 2000; c) Kuczkowski, A.; Fahrenholz, S.; Schulz, S.; Nieger, M. Organometallics 2004, 23, 3615; d) Schuchmann, D.; Kuczkowski, A.; Fahrenholz, S.; Schulz, S.; Flörke, U. Eur. J. Inorg. Chem. 2007, 931.
[2] a) Breunig, H. J.; Rösler, R. Coord. Chem. Rev. 1997, 163, 33; b) Breunig, H. J.; Ghesner, I. Adv. Organomet. Chem. 2003, 49, 95; c) Schulz, S. Adv. Organomet. Chem. 2003, 49, 225; d) Balázs, L.; Breunig, H. J. Coord. Chem. Rev. 2004, 248, 603.
[3] See for instance: a) Breunig, H. J.; Jawad, H. Z. Naturforsch. 1982, 37b, 1104; b) Breunig, H. J.; Jawad, H. J. Organometal. Chem. 1984, 277, 257; c) Breunig, H. J.; Krüger, T.; Lork, E. J. Organomet. Chem. 2002, 648, 209.
[4] a) Wieber, M.; Sauer, I. Z. Naturforsch. 1987, 42b, 695; b) Breunig, H. J.; Ebert, K. H.; Schulz, R. E.; Wieber, M.; Sauer, I. Z. Naturforsch. 1995, 50b, 735; c) Li, X.-W.; Lorberth, J.; Ebert, K. H.; Massa, W.; Wocadlo, S. J. Organomet. Chem. 1998, 560, 211; d) Breunig, H. J.; Königsmann, L.; Lork, E.; Nema, M.; Philipp, N.; Silvestru, C.; Soran, A.; Varga, R. A.; Wagner, R. Dalton Trans. 2008, 1831.
[5] Breunig, H. J.; Gülec, S. in B. Krebs (Ed.), Unkonventionelle Wechselwirkungen in der Chemie Metallischer Elemente, VCH, Weinheim, 1992, 218.
[6] a) Dehnert. P.; Grobe. J.; Hildebrandt. W.: Duc le Van. Z. Naturforsch. 1979, 34b, 1646; b) Wieber, M.; Sauer, I. Z. Naturforsch. 1984, 39b, 1668; c) Breunig, H. J.; du Mont, W. W.; Müller, D.; Severengiz, T. Z. Naturforsch. 1985, 40b, 848; d) Ashe, III, A. J.; Ludwig Jr., E. G. J. Organomet. Chem. 1986, 308, 289; e) Calderazzo, F.; Morvillo, A.; Pelizzi, G.; Poli, R.; Ungari, F. Inorg. Chem. 1988, 27, 3730.
[7] a) Heimann, S.; Schulz, S.; Bläser, D.; Wölper, C. Eur. J. Inorg. Chem. 2013, 4909; b) S. Heimann, D. Bläser, C. Wölper, S. Schulz, Organometallics 2014, 33, 2295.
[8] H. J. Breunig, H. Jawad, J. Organometal. Chem. 1984, 277, 257-260; c) H. J. Breunig, T. Krüger, E. Lork, J. Organomet. Chem. 2002, 648, 209-213.
[9] M. Wieber, I. Sauer, Z. Naturforsch. B 1987, 42, 695-698;
[10] a) P. Dehnert, J. Grobe, W. Hildebrandt, D. le Van, Z. Naturforsch. B 1979, 34, 1646-1652; b) M. Wieber, I. Sauer, Z. Naturforsch. B 1984, 39, 1668-1670; c) H. J. Breunig, W. W. du Mont, D. Müller, T. Severengiz, Z. Naturforsch. B 1985, 40, 848-849; d) A. J. Ashe, III, E. G. Ludwig, Jr., J. Organomet. Chem. 1986, 308, 289-296; e) F. Calderazzo, A. Morvillo, G. Pelizzi, R. Poli, F. Ungari, Inorg. Chem. 1988, 27, 3730-3733.
[11] a) H. J. Breunig, S. Gülec, Z. Naturforsch. B 1986, 41, 1387-1390; b) R. S. Dickson, K. D. Heazle, J. Organomet. Chem. 1995, 493, 189-197; c) S. Schulz, A. Kuczkowski, M. Nieger, J. Organomet. Chem. 2000, 604, 202-207.
[12] a) A. G. Davies, S. C. W. Hook, J. Chem. Soc. B 1970, 735-737; b) M. Wieber, U. Baudis, Z. Anorg. Allg. Chem. 1977, 436, 101-104; c) M. Wieber, U. Baudis, Z. Anorg. Allg. Chem. 1978, 439, 134-138; d) D. N. Kravtsov, B. A. Kvasov, S. I. Pombrik, E. I. Fedin, J. Organomet. Chem. 1975, 86, 383-395; e) G. G. Briand, A. Decken, N. M. Hunter, G. M. Lee, J. A. Melanson, Polyhedron 2012, 31, 796-800.
[13] T. Arnold, D. H. R. Barton, J.-F. Normant, J. Org. Chem. 1999, 64, 3722-3725.
[14] S. Heimann, A. Kuczkowski, D. Bläser, C. Wölper, R. Haak, G. Jansen, S. Schulz, Eur. J. Inorg. Chem. 2014, 28, 4858.