Project P5
Project description
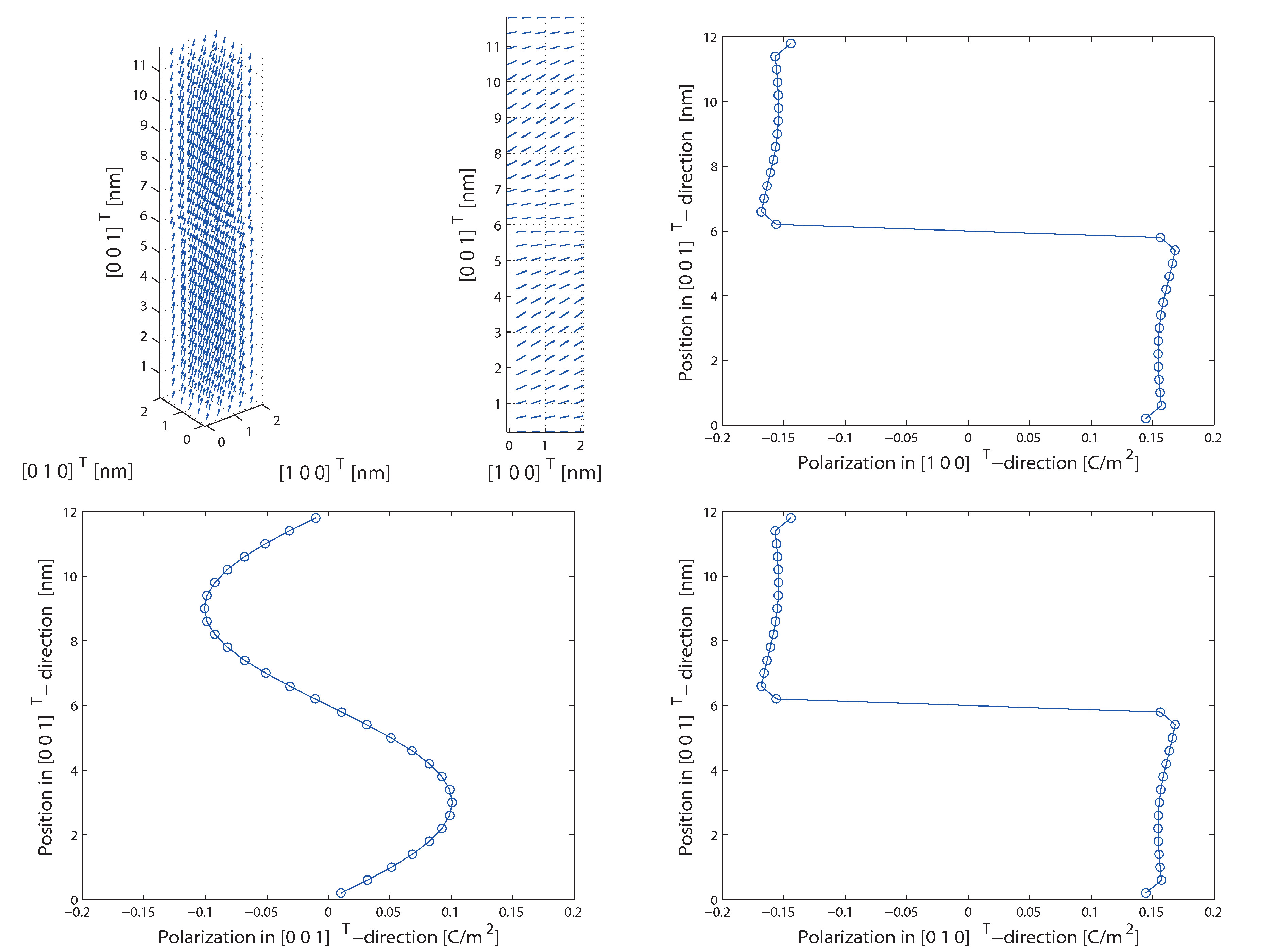
Molecular static methods for the simulation of ferroelectric materials
Ferroelectric materials are modelled and simulated on different length scales between the electron structure and the continuum description. Therefore, also atomistic models and algorithms have been developed further in the last decade and attain accurate results.Nevertheless the computational costs of the established atomistic methods are still significant. For that reason atomistic scale simulation methods are further developed.The goal of this project is the development and investigation of molecular static methods for the analysis of electromechanical phenomena on the micro level in ferroelectric materials. Therefore an accurate description of the material on an atomistic scale has to be made to investigate relevant regions, such as domain walls and nanofilms. In order to simulate ferroelectric materials on the micro scale, ab initio methods and molecular dynamics have been used for some time. Yet, not only the small length scale is a limiting factor but also the time step length. Through the use of molecular static methods the dynamic problem shall be transferred to a quasi-static algorithm.In order to simulate ferroelectric materials, the core-shell model is widely used in atomistic simulations. Thereby interactions between atom cores and electron shells are considered by interaction potentials, e.g. the Coulomb potential or the Lennard-Jones potential. The newly developed algorithm has been already used to investigate ferroelectric phenomena, such as domain walls, at the atomistic level. Moreover a new method has been implemented in order to consider macroscopic continuum stresses. Therefore the developed algorithm was able to compute not only the dielectric hysteresis but also the butterfly hysteresis of a ferroelectric crystal.Most molecular static algorithm do not consider temperature. However, in order to simulate phase transitions temperature has to be considered. Therefore a temperature dependent interaction potential has to be included to extend the core-shell model in order to simulate technically relevant structures and phase transitions.In addition to the targeted methodological outcome, the results shall be compared directly to the other projects which use non-atomistic methods for the simulation of ferroelectric materials.
Publications
Latest results

Prof. Dr.-Ing. habil. Paul Steinmann
Chair of Applied Mechanics
paul.steinmann@ltm.uni-erlangen.de
Tel.: +49-9131-8528501
Room: 0.037

Dipl.-Ing. Florian Endres
Chair of Applied Mechanics
florian.endres@ltm.uni-erlangen.de
Tel.: +49-9131-8528521
Room: 0.054
Vishal Boddu, M. Sc.
Chair of Applied Mechanics
vishal.boddu@ltm.uni-erlangen.de
Tel.: +49-9131-8564410
Room: 00.025